1. What is IPA? |
2. Description |
3. IPA Software
4. FAQ |
5. References |
6. Acknowledgement |
7. Summary
If you want to align protein sequences but you do not know what alignment parameter values to use, you can use IPA Software (or IPA for short) to find a good alignment parameter values to use. IPA takes biologically correct protein multiple alignments written in FASTA format (and optionally a substitution cost matrix in BLOSUM format) and gives a set of alignment parameter values which later you can use for aligning protein sequences. Currently, IPA finds gap-open cost, gap-extension cost and substitution cost matrix but other alignment parameters is under consideration. If you are curious what IPA gives, take a look at an (trimmed) example output:
# Generated by IPA v0.8 from examples specified in file(s) "*.fasta". # The minimizing average absolute error criterion is used. # The number of linear program variables considered is 416. # The number of iterations is 6. # The number of examples are 204. # The number of alignment parameters learned is 212. ## The gap-open cost is 42. ## The gap-extension cost is 22. A R N D C Q E G H I L K M F P S T W Y V 20 42 26 45 43 17 50 54 27 21 36 61 49 61 16 44 38 36 36 100 26 36 33 32 27 66 33 12 37 61 33 44 61 49 38 13 51 34 33 46 86 39 35 49 1 40 59 64 78 54 65 63 100 100 21 44 59 58 87 57 50 65 85 54 32 28 43 35 38 45 100 38 32 54 48 63 63 25 43 68 63 100 62 45 62 82 48 30 35 67 10 42 60 56 89 55 57 56 66 44 30 36 50 38 21 41 45 44 43 58 41 33 47 46 49 59 39 47 63 9 28 38 33 35 50 35 30 34 39 55 58 38 46 56 32 21 37 38 31 41 58 39 32 46 59 42 41 47 49 40 42 27 21 46 57 45 63 73 48 44 46 50 57 48 54 32 34 49 38 39 0 39 57 57 49 48 42 45 44 40 37 41 48 45 27 54 45 40 25 13 30 43 65 81 43 40 40 52 58 22 28 54 35 31 42 39 33 37 32 12 |
(Top)
IPA Software (or IPA for short) is a C language implementation of the algorithms of Kim and Kececioglu [KK06, KK07] for Inverse Parameteric Sequence Alignment. This is the following problem: Given a collection of examples of biologically correct alignments, finds alignment parameter values that make the cost of examples close to optimal.
IPA considers two types of examples: partial examples and complete examples. Partial examples are alignments consisting of reliable regions (or core blocks) interspersed with unaligned stretches. Complete examples are alignments with only reliable regions. The approach to partial examples builds upon a solution to the problem with complete examples.
The software also considers two criteria for measuring closeness to optimality. In finding parameters that make the cost of the examples close to the cost of an optimal alignment of the example strings, the error in cost is optimized by minimizing either the maximum relative error or the average absolute error of examples.
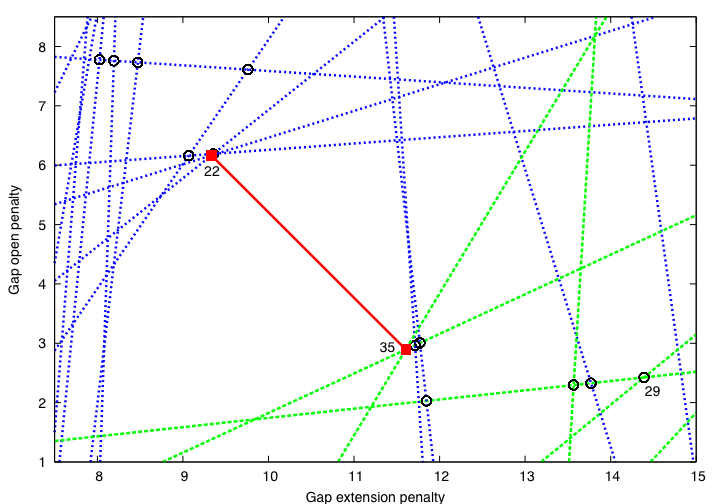
Inverse parametric sequence alignment is solved by a reduction to linear programming. The software solves the resulting linear program by a cutting plane algorithm, which iteratively adds inequalities to a linear program. The following figure, from [KK06], illustrates this approach on a simplified instance that finds the two affine gap penalties for a fixed substitution matrix.
(Top)
IPA is a system independent implementation and runs on major operating systems once it's been compiled and a binary image is produced on your system. Currently IPA is distributed as a source code package. Hence you need to compile the package first. To compile IPA you need a working C compiler with its standard C library and a working GLPK library on your system. GLPK (short for GNU Linear Programming Kit) is intended for solving large-scale linear programming (LP), mixed integer programming (MIP), and other related problems. It is a set or routines written in ANSI C and organized in form of a callable library. It is a freely distributed by GNU under GNU General Public License.
If you are using a UNIX-based system such as Linux, BSD, and OS X, and familiar with compiling source packages on your system, you can obtain a source copy of the latest GLPK at http://www.gnu.org/software/glpk/. If you prefer installing binary packages, there are RPM or DEB packages you can use. Check the major package distribution sites. If you use a Mac OS X system, you can download GLPK binary package from Fink at http://finkproject.org/. If you are not familiar installing source or binary packages, ask your system administrator or someone who is an expert in system management around you. Without GLPK library installed, IPA won't be compiled.
(Top)
You can obtain IPA source code distribution on this website via electronic download (and that's the only way to obtain the distribution at this time). But before you do, you should read "IPA Software Terms of Use" and accept it first.
| IPA Software is free for noncommercial use. IPA Software comes with no warranty nor guarantee; one may use it at his own risk. IPA cannot be redistributed in any form. If one wishes to use IPA Software for commercial purpose, she should obtain the permission from each author first. By downloading any of distribution package, he accepts these terms. (end of terms) |
Once you accept "IPA Software Terms of Use" you can download the latest version of IPA source code distribution by clicking one of the following links.
Make sure that you download the most recent version of the software if there's more than one version. Once you finish downloading, you need to compile it to obtain a binary image running on your system.
(Top)
Please make sure that your system meets the requirement. To compile the source code, you need to unpack the downloaded file. If you do not know how to unpack it, please type, in the directory where the file resides, either one of the following two on the command-line prompt depending on the format you chose to download.
Once you've compiled the source code successfully, you should have a binary image called ipa. Please verify if it's running on your system and its version number is the same as x.y by typing ipa --version.
(Top)
ipa works with a few options. For now, the most important option is --help. Type
ipa --helpto see what options ipa accepts. You can also read an online manual page here. Although there are many way to control the behaviour of ipa, most of time you don't need to provide any options. But you always need to give at least one multiple alignment file in FASTA format to ipa. If you have protein multiple alignment files in CLUSTAL format, here's a small perl script called clustal2fasta.pl which converts them into FASTA format. To see how it works, type clustal2fasta.pl --help.
We show some examples of IPA usage. First download dataset PALI.fasta which contains five protein multiple sequence alignments in FASTA format. Once you finish downloading, IPA can learn from these multiple alignments to obtain different sets of alignment parameter values using different methods. Try the followings one by one.
(Top)
If you find any bug or typo in IPA Software (including documentation as well as source code) or in this website, please contact us via email shown at the end of this page. We appreciate your reports.
(Top)
(Under construction.)
(Top)
Below is the list of papers on the algorithms behind IPA. To refer to IPA in a paper, please cite reference [KK06] below.
(Top)
This research was supported by the US National Science Foundation through Biological Databases and Informatics Program Grant DBI-0317498, "Robust Tools for Biological Sequence Alignment."
(Top)
| URL of this page | http://inversealign.cs.arizona.edu | |
| IPA Software Terms of Use | Please read it and accept it before you start downloading the distribution. | |
| Release version | 1.0 | |
| Distribution |
IPA-1.0.tar.gz IPA-1.0.tar.bz2 |
|
| Documentation | The distribution contains all documentation, including this online manual. | |
| Citation |
Kim, E. and J. Kececioglu,
"Inverse sequence alignment from partial examples,"
Algorithms in Bioinformatics, 7th International Workshop (WABI)
359—370, 2007. (PDF) Kececioglu, J. and E. Kim, "Simple and fast inverse alignment," Proceedings of the 10th ACM Conferences on Research in Computational Molecular Biology (RECOMB) 441—455, 2006. (PDF) |
|
| Funding |
US National Science Foundation Grant DBI-0317498, "Robust Tools for Biological Sequence Alignment." |
|
| Contact | inversealign@cs.arizona.edu |
(Top)
If you have suggestions (!) or questions (?), please contact us at
inversealign@cs.arizona.edu
http://inversealign.cs.arizona.edu
Updated on 8 April 2007
Take a look, a Wikipedia definition of IPA ;^)